
Autophagy can be Fun!
Or at least not boring and confusing!

Welcome back to an aging blog – now that I’ve finished my digression into cholesterol, I can get back to my obsession with the molecular and cellular mechanics of aging. Here, I’m going to go into a pretty obscure topic – but one that I think may grow into a big and important area of not just aging research, but relevant to a whole host of other diseases.
I started down this rabbit hole when I read about the work of a physician/scientist from Harvard, Anna Greka, who works on rare genetic diseases. These are typically caused by mutation(s) in a single gene. If you want the overview, she has a great TED talk.
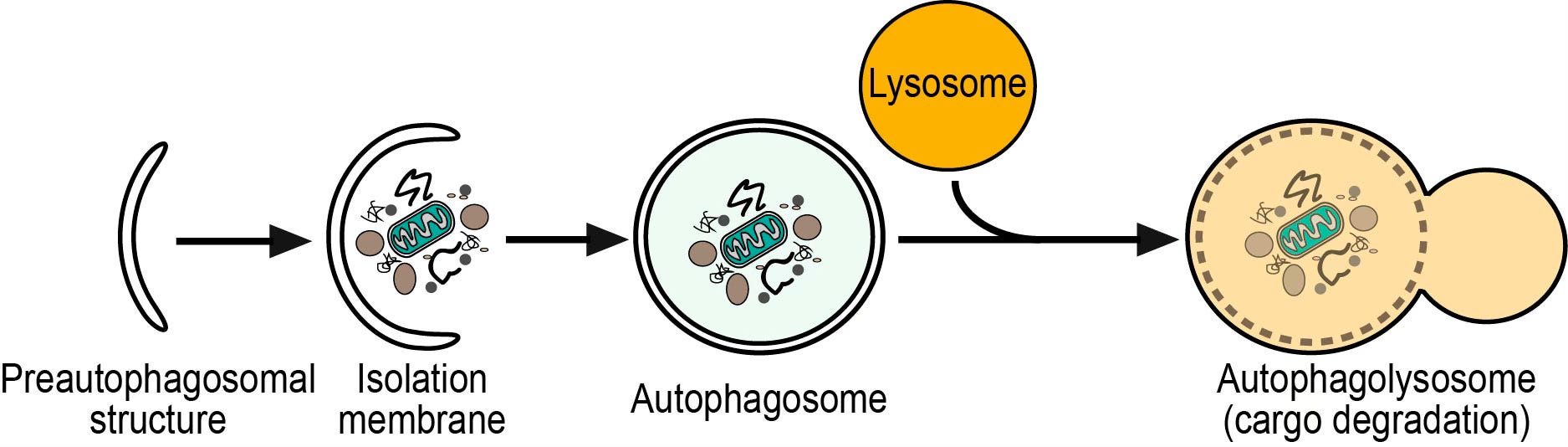
Before I get to her fascinating finds, there is, as usual, a back story, this one on autophagy. I’ve written on this very cool process before, as this cellular recycling capability is turned up by intermittent fasting and other forms of caloric restriction. Things get a little complicated now, as there are different types of autophagy. The stress-induced, or starvation form of autophagy, is triggered, as implied, by lack of nutrients. It makes sense that cellular recycling would scavenges goodies from trash and offset nutrient deprivation. The better-studied, and continuously-occurring form is called selective autophagy. I’ll explain this below, but visuals can help clarify the process:
For a more simplistic overview:
For a deep dive with beautiful graphics (you can ignore molecular details and acronyms)
Keep in mind that cells, like the body in general, strive to maintain homeostasis, i.e. a constant and regulated environment. To do this, damaged, superfluous, and harmful material has to be removed. Selective autophagy is the main way cells do this. The junk that’s slated for removal is moved into structures called lysosomes, which contain enzymes that can break down different types of molecules. The component pieces can then either be re-used by the cell or transported out for elimination.
Selective autophagy removes a variety of unwanted materiel from cells. One group includes damaged organelles, such as mitochondria and parts of the endoplasmic reticulum (the manufacturing regions of the cell). Proteins that we can generically term mis-formed are also removed. These include proteins that have clumped together (aggregates), or have not passed the quality control checks following their synthesis. Finally, bacteria and viruses can be eliminated in this way.
The material the cells want to get rid of is tagged with a variety of labels that say ‘take me’, and put into nascent ‘sacs’ that form around it, eventually becoming a complete package called an autophagosome (literal translation: a self-eating body). The autophagosome is moved to and fused with a lysosome. You can see this process in the preceding videos. I want to make two points by stressing this rather obscure part of the autophagy process.
First, stuff the cell is getting rid of, the technical term is ‘cargo’ (!), is recognized by specific receptors in the cytoplasm (the liquid matrix in a cell). If you watched one of the videos, these are some of the many acronyms in the early stages of autophagosome formation. These receptors play a crucial role in the eventual degradation of cargo by forming clusters, or globs of the same or similar cargos. I’ll come back to this point.
Second, once labelled cargo is bound to a receptor, this initiates the process of building the enclosure around it: the autophagosome. The overview from the videos is useful in conceptualizing this process – it’s pretty complicated, involving numerous steps. My reason for emphasizing this is because if any one of these is blocked or doesn’t work right, the fusion of the sac with the breakdown apparatus in the lysosome won’t happen.
A failure here can result in the various diseases associated with defects in selective autophagy, perhaps most notably in the context of neurodegenerative diseases including Parkinson’s disease and tauopathies, but also the previously mentioned genetic diseases.
Let’s go back to misshapen proteins that are targeted for removal. Remember that proteins are big molecules, consisting of hundreds or thousands of amino acids. These chains are built from instructions in our DNA, which are copied into a similar molecule (RNA) that is decoded by a molecular machine called a ribosome.
For a brief, graphic refresher on protein synthesis you can watch this video.
Most proteins assume a 3D shape that facilitates their specific function(s) and this ‘folding’ as it is called, takes place after the ribosomal synthesis. Amazingly, about one-third of all newly synthesized proteins, especially long and/or difficult-to-fold proteins, fail to pass the quality control system that ensures proper folding. Normal protein sequence is disrupted by mutation, which means that normal folding won’t occur.
Another type of misfolded protein burden occurs in aging cells, where our QC system is less efficient. These damaged proteins contribute to diseases such as Parkinson’s and Alzheimer’s.
Back to Anna Greka’s work. She and her team found that one of the proteins (called TMED) involved in the earliest process of autophagy, recognizes and binds to a mutant protein (MUC1). Normally, this binding would trigger subsequent stages of formation of the autophagosome (the transport sac that carries damaged cargo to the lysosomes for disposal). The first of those stages involves moving the TMED bound to its cargo to the growing sac membrane. This is the normal aggregation step between cargo and transporter.
In the case of MUC1, however, this binding triggers a multiplier process. The MUC1 attached to the first TMED gloms on to the MUC1 attached to a 2nd TMED. This in turn gloms on to a third and on and on, eventually gumming up the whole works. Not only is the cargo not disposed of but a new and dangerous conglomerate structure disrupts the normal autophagy process. (A beautiful but complicated diagram of this process can be seen in Figure 6 of Greka’s paper).
In the case of the kidney disease Prof. Greka studies, the aggregate eventually killed the kidney cells. A similar process has been documented in retinitis pigmentosa, a form of blindness. Remarkably, based on the identification of the molecules involved in the aggregation process, the research group was able to find and test a treatment that reversed it in mice and test tubes by displacing the bound MUC1 and targeting it to the lysosome for breakdown.
Diseases caused by mutant proteins like the MUC1 are generically termed proteinopathies, i.e. pathologies due to a protein. Most of these are not exactly household names, such as cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy, neonatal diabetes caused by insulin gene mutations, and Ehlers-Danlos syndrome caused by collagen mutations.
These rare, so-called orphan diseases (because not many researchers study them due to their low numbers), could be amenable to treatment like that for MUC1. But even more exciting to me is the possibility that other diseases caused by protein aggregation could be reversed by this type of research and treatment. These include many neurodegenerative diseases such as Alzheimer’s, Parkinson’s, Huntington’s, and ALS. Other organ systems suffer from diseases caused by the abnormal accumulation of gobbed up proteins; these include type 2 diabetes and several heart diseases. There are a lot of years and potential mis-steps between bench (i.e. lab study) and bedside (i.e. clinical treatment), but this is promising and exciting work. I hope research funding for these groups is not getting cut.